![]()
Caspases
What Makes a Cell Commit Suicide?
Cells are receptive to both positive and negative growth signals. Cells require continuous stimulation from neighbouring cells in order to survive, such as the binding of growth factors for neurons. Negative growth signals, such as tumour necrosis factor-a (TNFa) or the Fas ligand (FasL), can curb survival. The delicate balance between positive and negative cell growth signals can shift, which can cause a cell to enter apoptosis.
Apoptosis follows two major pathways: one is extrinsic, relying on the stimulus of cell surface receptors such as Fas or TNF (TNFR), while the other is intrinsic, relying on the stimulus of mitochondrial receptors as a consequence of cellular stress or DNA damage.
Extrinsic Pathway
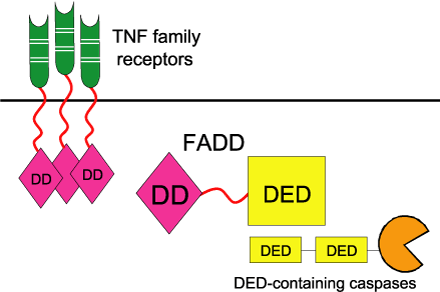
Specific cells, such as cytotoxic T cells, produce death activators that act upon cells selected to die. These death activators, such as FasL, TNFa or lymphotoxin, bind to receptors on the surface of the targeted cell, thereby triggering the activation of the cell death machinery. These receptors include TNF receptors that possess a Death domain, such as Fas and TNFR-6. The binding of a death activator (such as FasL) to its TNF receptor (such as Fas) activates the receptor. The activated receptor then transmits the apoptotic signal to the cytoplasm by recruiting FADD (Fas-Associated Death Domain protein) via its cytoplasmic Death domain, to form the death-inducing signalling complex (DISC). FADD contains two domains: a Death domain that binds to the Death domain on the Fas receptor, and a DED (Death effector domain) that binds to DED on pro-caspase-8. The proteolytic activation of caspase-8 leads to its dissociation from the DISC complex. Active caspase-8 can then initiate the caspase cascade that leads to apoptosis and phagocytosis via the proteolytic activation of other caspases, including caspases-3, -4, -6, -7, -9 and -10.
|
|
Linking TNF family death receptors to DED family caspases through the bipartite adapter FADD. The DD of FADD binds to the DD in the cytosolic tails of TNF family receptors. The DED of FADD binds the DEDs of pro-caspases 8 and 10. Reprinted
from Science’s STKE Jun 22(239), J.C. Reed, K.S. Doctor and A. Godzik, The
Domains of Apoptosis: a Genomics Perspective, ppRE9, 2004, PMID:
15226512 |
Intrinsic or Mitochondrial
Pathway
A second apoptotic mechanism arises from signals that originate from within the cell as a consequence of cellular stress or DNA damage, and which involves mitochondrial-signalling events. Bcl-2 family members are critical regulators of mitochondrial-dependent apoptosis, whereby pro- and anti-apoptotic family members control the permeability of the mitochondrial outer membrane to apoptogenic proteins. The initial events are thought to be the down-regulation of the anti-apoptotic mitochondrial membrane protein Bcl-2, and the activation of the pro-apoptotic Bcl-2 family member, Bax. Bax is activated directly by the p53 tumour suppressor protein following stress induction, or indirectly through the p53-activation of the Bcl-2 pro-apoptotic members Noxa and PUMA, or thorough p53-independent mechanisms. The activation of Bax results in its movement from the cytosol to the mitochondrial membrane, where it oligomerises and embeds itself in the membrane to create a pore. The down-regulation of Bcl-2 is required to prevent it blocking Bax oligomerization. The formation of the Bax pore, as well as the consequent loss of mitochondrial membrane potential, causes cytochrome c to leak out of the mitochondria into the cytosol. Once in the cytosol, cytochrome c forms a complex with Apaf-1 (apoptosis protease activating factor) and caspase-9 called the apoptosome. The Apaf-1 activation of caspase-9 within the apoptosome is a key event that triggers the activation of the caspase cascade, including caspases-3 and –7, which execute the cell death programme.
The final result of both pathways is the caspase-activation of proteins involved in the destruction of the cell’s contents. For example, apoptotic endonucleases cleave the DNA between nucleosomes, resulting in the fragmentation of DNA. One such endonuclease is CAD, which contains a CIDE domain and is held in an inactivate state by the CIDE domain-containing chaperone protein, ICAD, through CIDE-CIDE interactions. The cleavage of the ICAD chaperone by caspases releases an activated CAD endonuclease that can then digest the DNA.
Next: The Domains of Apoptosis
Previous: Caspases: Key Apoptotic Proteins