
Amyloid-b Precursor Protein
Amyloid
Plaque Formation
|
|
|
APP Processing: a-secretase and g-secretase produce non-plaque forming p3, while b-secretase and g-secretase produce amyloid plaque-forming Ab. The different regions of the APP protein are indicated. |
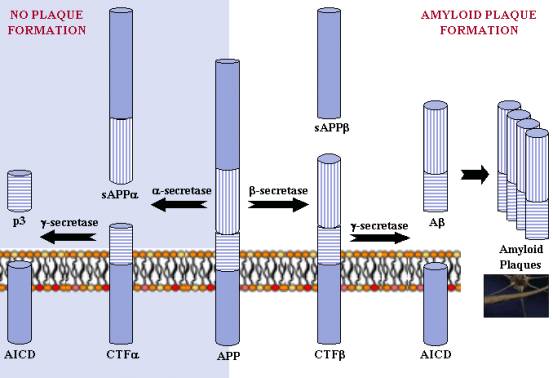
APP can be processed in different ways by different sets of enzymes - one pathway leads to amyloid plaque formation (amyloidogenic), while another does not (non-amyloidogenic). Usually about 90% of APP enters the non-amyloidogenic pathway, and 10% the amyloidogenic one, but these ratios can change due to mutations, environmental factors, as well as the age of the individual. Cleavage products from both these pathways may play important roles in neural development and function.
Non-Plaque-forming (non-amyloidogenic) Pathway
In the non-plaque-forming pathway, APP is cleaved first by a-secretase to yield a soluble N-terminal fragment (sAPPa) and a C-terminal fragment (CTFa). sAPPa may be involved in the enhancement of synaptogenesis, neurite outgrowth and neuronal survival, and are considered to be neuroprotective. CTFa is retained in the membrane, where it is acted upon by presenilin-containing g-secretase to yield a soluble N-terminal fragment (p3) and a membrane-bound C-terminal fragment (AICD, or APP intracellular domain). AICD may be involved in nuclear signalling via transcriptional regulation as well as axonal transport through its ability to associate with a host of different proteins.
Plaque-forming (amyloidogenic) Pathway
In the plaque-forming pathway, APP is cleaved first by a different enzyme, b-secretase (a transmembrane aspartic protease), yielding a soluble N-terminal fragment (sAPPb) and a membrane-bound C-terminal fragment (CTFb). This cut is made closer to the N-terminal end of APP than with a-secretase, making CTFb longer than CTFa. CTFb is then acted upon by g-secretase (as occurred in the previous pathway), yielding a membrane-bound C-terminal fragment (AICD) the same as before, and a soluble N-terminal fragment (amyloid-b, or Ab) that is longer than p3. Though Ab is required for neuronal function, it can accumulate in the extracellular space of the brain, where it can aggregate to form amyloid plaques. Ab can exert deleterious effects on neuronal and synaptic function, ultimately causing neuronal cell death.
Plaque Formation
Ab is ‘stickier’ than other APP fragments, and accumulates by stages into microscopic plaques. Plaques form by a multi-step polymerisation mechanism, with Ab peptides aggregating into oligomers, which cluster together to form fibrils with a regular b-sheet structure. These fibrils adhere together to form mats, which clump together with other substances to finally form plaques. Ab plaques may disrupt brain cells by clogging points of cell-cell communication, as well as activating immune cells that trigger inflammation, which can be lethal to cells. In addition, Ab is thought to cause oxidative damage to cells.
Alzheimer’s Disease
Ab is not all bad. Soluble Ab oligomers are required for Ab-induced inhibition of long-term-potentiation. However, the insoluble amyloid plaques appear to disrupt neuronal function. The problem may have to do with the aggregation state of the peptide, which in turn may be influenced by the concentration, structure and length of Ab peptide present.
The concentration of Ab in the extracellular space may affect its toxicity by influencing its aggregation. Ab concentration appears to be influenced by several factors, including the level of neural activity and synaptic release, which seem to increase Ab levels. Age could also play a factor, studies showing that the level of a-secretase decreases with age, while b-secretase activity appears to increase. In addition, studies indicate that cholesterol may play a role in regulating Ab production, with high levels of cholesterol being linked with increased Ab release and plaque formation. One possible explanation for this result is the presence of cholesterol-rich regions of membranes known as lipid rafts, which may affect the distribution of APP-cleaving enzymes.
The structure of the Ab oligomers could influence plaque formation. Whereas soluble monomeric Ab peptide has been shown to have an unordered structure in solution, within amyloid fibrils Ab adopts a regular b-sheet conformation. The conformational change to a b-sheet structure may trigger the formation of the abnormal fibrillar amyloid plaques found in AD patients.
Ab peptides can differ in length from 38 to 42 amino acids. Ab ending at amino acid 42 (Ab42) appears to be the main species of Ab peptide deposited in plaques. In AD patients, studies show an increase in the ratio of Ab42 to Ab40. In addition, genetic changes involved in familial AD result in increased amounts of Ab42 present in the brain. Ab peptides are metal chelators with the ability to reduce those metals. For example, in vitro, Ab can reduce Cu2+ to Cu+, and Fe3+ to Fe2+. Ab42 appears to be a more effective reductant than Ab40, which may contribute to the ability of Ab42 to activate mononuclear phagocytes in the brain and elicit an inflammatory response.
Therefore, it appears that there may be several contributory factors to plaque formation in AD patients, the relative importance of which still need to be deciphered.
Next: What InterPro Tells Us
Previous: Amyloid-b Precursor Protein, Still and Enigma