Alphafold, A Novel Tool Shedding Light On Alzheimer’s Disease Proteins
focusAlzheimer’s disease (AD) is the most common type of dementia, affecting around 50 million patients worldwide. It has been present from the early time of mankind: the ancient Egyptians, around 2000 BC, were aware of memory declines in their population. However, it was not until 1906 that the German psychiatrist Alois Alzheimer reported the first case of this pathology [1, 2]. It is a multifactorial, irreversible, progressive neurodegenerative disorder characterised by memory loss and cognitive impairment that affects the language, personality and behaviour, losing the ability to execute simple everyday tasks [3, 4].
The neuropathology of AD is complex and the hallmarks are the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles (NFTs) in various areas of the brain, explained, mainly, by the amyloid cascade hypothesis. This hypothesis states that neurodegeneration is caused by abnormal accumulation of misfolded amyloid beta (Aβ) peptides into plaques which act as a trigger for a cascade that includes formation of NFTs (abnormal filaments of hyperphosphorylated tau protein that mediate the disintegration of microtubules in brain cells) and activation of different pathways leading to inflammatory response and oxidative stress. This cascade results in neuron damage, loss of synapses and neurons, with the consequent cognitive impairment and cortex atrophy [5, 6, 7].
Aβ peptides derive from amyloid precursor protein (APP, IPR028866), an integral membrane multidomain protein of 695-770 amino acids, as illustrated in Figure 1 and 4, involved in several functions including signalling, neuronal development and neural plasticity, antimicrobial activity, iron transport and anterograde transport. It can be post translationally processed by two pathways: via a non-amyloidogenic pathway through alpha and beta-secretases cleavage to generate monomeric Aβ (IPR013803) implicated in diverse biological functions such as memory, learning, and neuroprotection [5, 6, 7]; or via an amyloidogenic pathway by beta and gamma-secretases, generating fragments of different size, further cleaved to the main Aβ forms of 40 and 42 amino acids which aggregate into insoluble amyloid plaques.

Figure 1. Domains of human APP (P05067) in InterPro. From N- to C-terminus: Heparin-binding domain (IPR015849), copper-binding domain (IPR011178), Kunitz domain (IPR002223), E2 domain (IPR024329), Aβ peptide (IPR013803) and C-terminal domain (IPR019543).
The complete crystal structure of APP has not been resolved, but only for domains and short Aβ fragments. Determination of protein 3D structure is laborious and time consuming, thus bioinformatic tools are key. Recently, DeepMind in conjunction with EMBL-EBI launched AlphaFold DB based on the results of AlphaFold 2.0, an AI system that predicts highly accurate 3D protein structures from the amino acid sequence for the human proteome and 20 other key organisms. This is particularly useful for proteins without an experimental structure, such as APP [8, 9]. In InterPro we have integrated these structure predictions for protein and InterPro entry pages, when available, with a link to AlphaFold DB. The 3D viewer includes a colour-coded reference for the per-residue prediction confidence, therefore, the bluer the colour the higher the confidence, as shown in Figure 2.
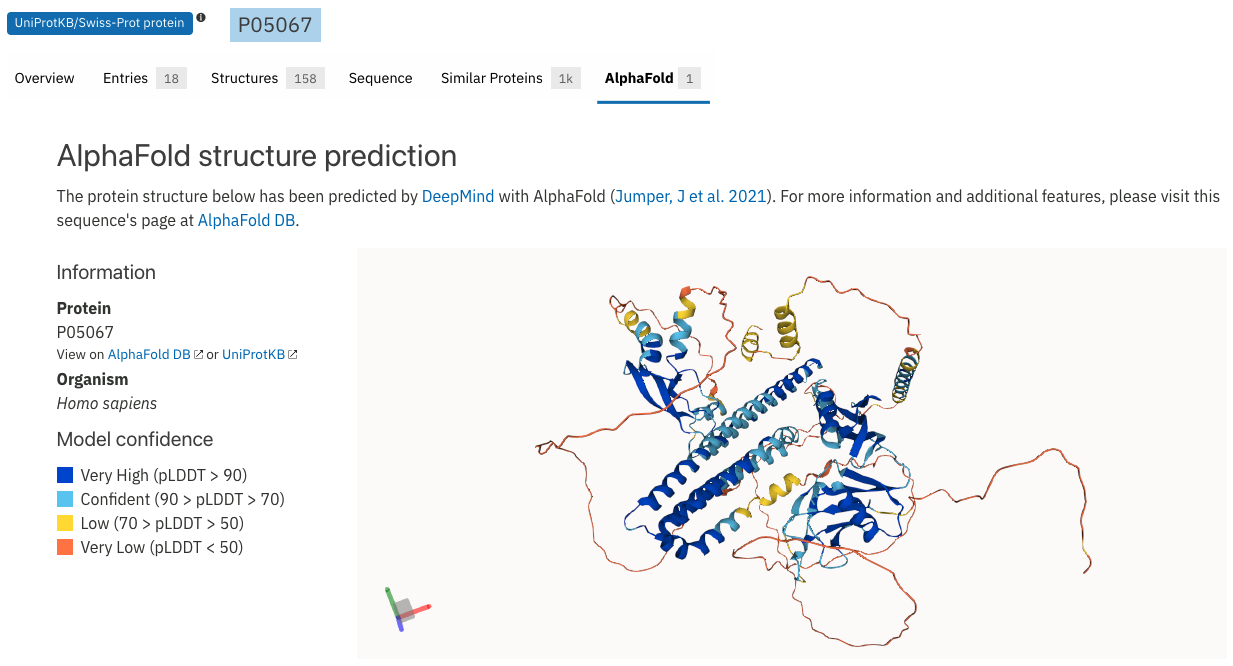 Figure 2. AlphaFold structural model for human APP (P05067).
Figure 2. AlphaFold structural model for human APP (P05067).
The AlphaFold page also includes a predicted aligned error section which is useful to assay accuracy between domains, in which darker green represents lower error of the prediction (Figure 3). In the case of the human APP, the structure prediction for individual domains correlates very well with the structural information available. Note that the segment from which Aβ derives (between 670-710) is mostly unstructured. However, this orientation of the different regions has not been confidently predicted by AlphaFold. The relative positions for the heparin-binding and the copper-binding domains seems to be more confidently predicted than the other domains as indicated by the dark green shown beside the light pink and magenta squares in Figure 3.
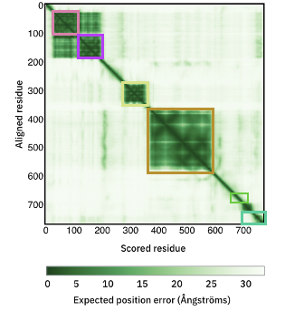
Figure 3. Predicted align error per domain in AlphaFold structural model of human APP (P05067). Squares highlight domains as in Figure 1: Heparin-binding domain in light pink, copper-binding domain in magenta, Kunitz domain in lime, E2 domain in light brown, Aβ peptide in green and C-terminal domain in light-cyan.
Figure 4 shows the AlphaFold structural model APP (P05067), with domain organisation in the full length protein sequence, coloured as in Figure 1.
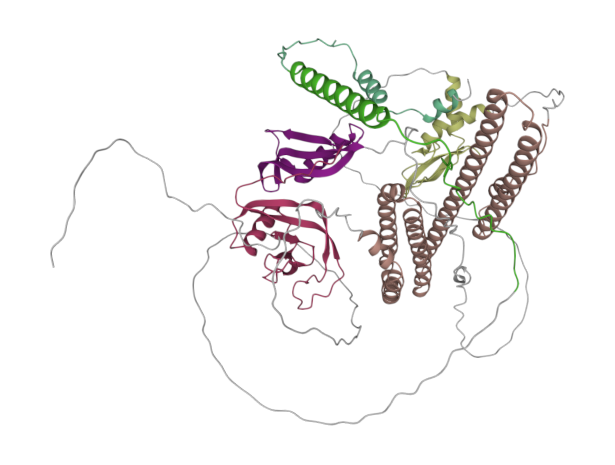 Figure 4: Predicted domains in AlphaFold structural model for human APP, displayed using Molstar [10].
Figure 4: Predicted domains in AlphaFold structural model for human APP, displayed using Molstar [10].
For the unstructured Aβ peptide, MD simulations, confirmed by amyloid-oligomer-specific antibodies, revealed that Aβ monomer (Figure 5A) acquires the atypical α-sheet secondary structure that adopts an α-strand structure (Figure 5B) which proceeds to an α-sheet between adjacent α-strands in oligomers with opposite charges on both edges, inducing self-assembly/aggregation to form soluble oligomeric amyloid protofibrils (Figure 5C) and, finally, insoluble highly ordered amyloid fibrils with a cross β-sheet structure (Figure 5D) [5-7]. MD simulations, confirmed by amyloid-oligomer-specific antibodies, unveil the possible mechanism of Aβ aggregation.
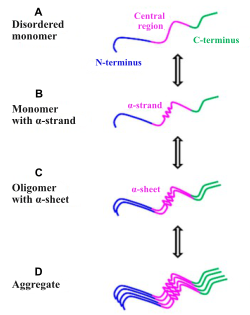 Figure 5. Image extracted from Aggregation Mechanism of Alzheimer’s Amyloid β-Peptide Mediated by α-Strand/α-Sheet Structure [5].
Figure 5. Image extracted from Aggregation Mechanism of Alzheimer’s Amyloid β-Peptide Mediated by α-Strand/α-Sheet Structure [5].
This is a good example in which computational structure predictions are key tools in research, particularly in the case of membrane proteins that are difficult to crystallize, such as APP. These predicted models, in the context of the whole protein, are extremely useful to refine domain boundaries of, for example, Pfam families, providing more accurate annotations both in Pfam and InterPro databases. Hopefully, these tools, combined with experimental techniques, may contribute to the understanding of the pathological mechanisms that lead to protein aggregation and toxicity in AD development, and therefore, support the drug design field to find effective treatments, especially to target the early events of AD pathogenesis to delay its progress.
References:
- Yang HD et al. History of Alzheimer’s Disease. Dement Neurocogn Disord. 2016 Dec;15(4):115-121. (PMID:30906352).
- Breijyeh Z et al. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules. 2020 Dec 8;25(24):5789. doi: 10.3390/molecules25245789. (PMID:33302541).
- Soria Lopez JA et al. Alzheimer’s disease. Handb Clin Neurol 2019;167:231-255. doi:10.1016/B978-0-12-804766-8.00013-3. (PMID:31753135).
- Chen GF et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017 Sep;38(9):1205-1235. doi: 10.1038/aps.2017.28. Epub 2017 Jul 17. (PMID:28713158).
- Balupuri A et al. Aggregation Mechanism of Alzheimer’s Amyloid β-Peptide Mediated by α-Strand/α-Sheet Structure. Int J Mol Sci. 2020 Feb 7;21(3):1094. doi: 10.3390/ijms21031094. (PMID:32046006).
- Shea D et al. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2019 Apr 30;116(18):8895-8900. doi: 10.1073/pnas.1820585116. Epub 2019 Apr 19. (PMID:31004062).
- Li D et al. Structural Diversity of Amyloid Fibrils and Advances in Their Structure Determination. Biochemistry. 2020 Feb 11;59(5):639-646. doi: 10.1021/acs.biochem.9b01069. Epub 2020 Jan 28. (PMID:31967790).
- Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021 Jul 15;1-7. doi: 10.1038/s41586-021-03819-2 (PMID:34265844)
- Tunyasuvunakool K et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021 Jul 22. doi: 10.1038/s41586-021-03828-1 (PMID:34293799).
- D.Sehnal et al. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures, Nucleic Acids Research, 2021; doi: 10.1093/nar/gkab31 (PMID:33956157).